 Weghorn Lab
Weghorn Lab
Computational Biology and Health Genomics
- Group page
- Research lines
- Group members
- Publications
- Datasets
- Software


October 2018 Group Leader in the Bioinformatics and Genomics Programme at the Centre for Genomic Regulation, Barcelona, Spain.
2013-2018 Postdoctoral researcher at the Department of Medicine and Department of Biomedical Informatics, Harvard Medical School, Boston, USA
2012 PhD in Theoretical Physics, University of Cologne, Germany
Group News
No evidence for a controversial type of mutation bias in the human germline (08/07/2020)
Recent studies have found that some human cancer cells display a mutation bias where DNA that directly codes for proteins mutates at a different rate compared to DNA sequences which do not.
A comprehensive list of cancer driver genes published in Nature Genetics (04/02/2020)
An innovative algorithm identifies 460 genes that are important for the development of cancer, uncovering tumor-gene associations that had not previously been identified.
Summary
Mutation, selection and stochasticity combine to leave their footprint in genetic data. With the proper tools and analyses, the resulting information can be leveraged to describe mutational processes, obtain estimates of selection, and reveal more intricate evolutionary dynamics. We are interested in the quantitative description of evolutionary processes through the development of new probabilistic models and computational methods.
A strong focus of the lab lies on cancer as an evolutionary system and selection as a readout. We analyze sequencing and other datasets and develop mathematical and computational approaches to estimate selection. An important aspect of this effort is to account for the high heterogeneity of mutation rate in cancer genomes, driven by sequence context dependence and external mutation rate-determining factors. We are interested in different modes of selection active during tumorigenesis (including negative selection), happening in the coding or the noncoding part of the genome. We extend some of the analyses to the level of human population genetics, which is another active area of research in our lab.
External lab website: weghornlab.net
Job Openings
We are always interested to receive inquiries from researchers at any level who are excited about quantitative and computational science and would like to work in the fields of cancer evolution or human population genetics. Please get in touch with Donate Weghorn.
Funding Acknowledgements

The project “ESTIMACION PRECISA DE LOS PARAMETROS PARA LA INFERENCIA DE LA SELECCION EN LOS GENOMAS DEL CANCER HUMANO” (PID2021-128976NB-I00_DWeghorn) is funded by Agencia Estatal de Investigación (AEI), the Ministerio de Ciencia e Innovación and Fondo Europeo de Desarrollo Regional (FEDER). Project PID2021-128976NB-I00_DWeghorn funded by MCIN/ AEI / 10.13039/501100011033 / FEDER, UE
![]()
The project 'TAFI (Tumor Allele Frequency Interpreter) : una eina de deep learning per la classificació de tumors', granted under the call from 'Industria del Coneixement (Llavor i Producte), has received funding from the Department of Research and Universities, of the Government of Catalonia
Period: 12/10/2022 to 11/07/2023
Cancer evolution
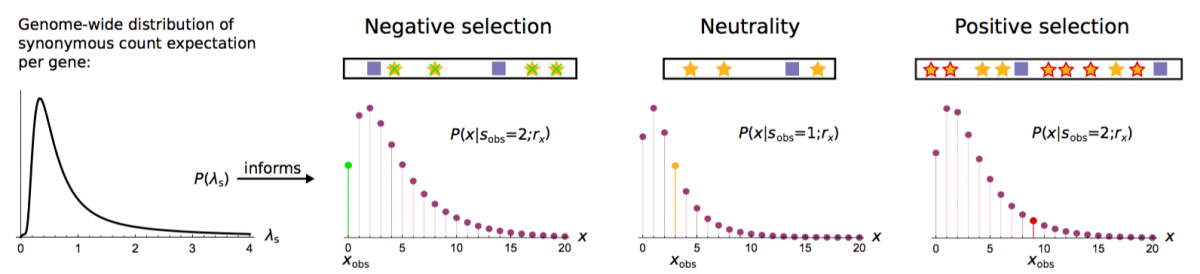
- Dietlein*, F., Weghorn*, D., Taylor-Weiner, A., Richters, A., Reardon, B., Liu, D., & Sunyaev, S. R. (2020). Identification of cancer driver genes based on nucleotide context. Nature Genetics, 52, 208-218. Journal bioRxiv
- Weghorn, D. & Sunyaev, S. (2017). Bayesian inference of negative and positive selection in human cancers. Nature Genetics, 49, 1785. Journal SharedIt PubMed
- D'Antonio*, M., Weghorn*, D., D'Antonio-Chronowska, A., Coulet, F., Olson, K. M., DeBoever, C., Drees, F., Arias, A., Alakus, H., Richardson, A. L., Schwab, R. B., Farley, E. K., Sunyaev, S. R. & Frazer, K. A. (2017). Identifying DNase I hypersensitive sites as driver distal regulatory elements in breast cancer. Nature Communications, 8(1), 436. Journal
Human population genetics
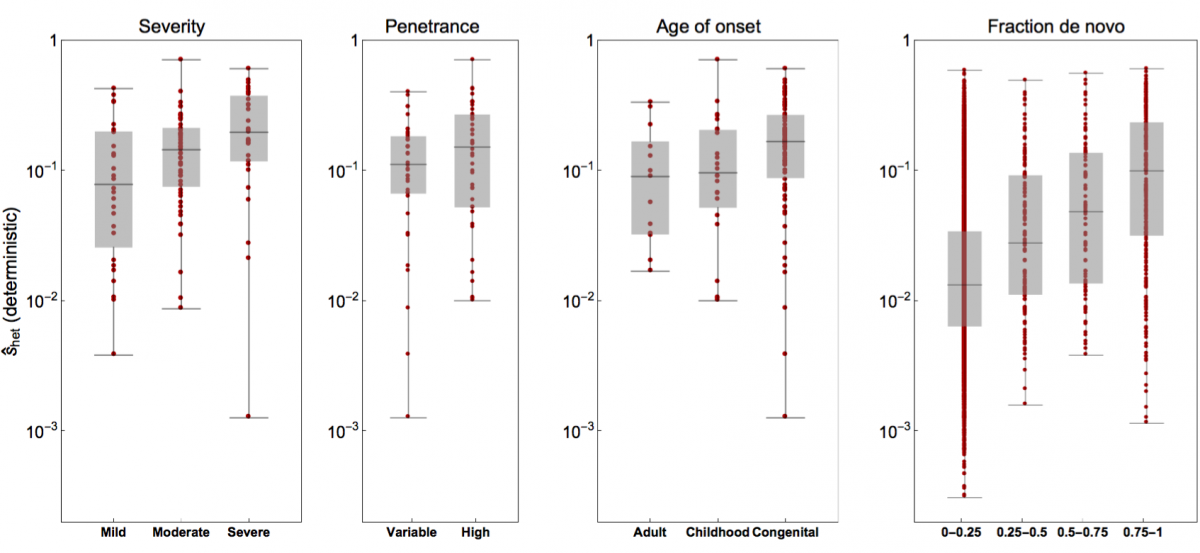
- Rodriguez-Galindo, M., Sònia Casillas, Weghorn†, D. and Barbadilla†, A. (2019). Germline de novo mutation rates on exons versus introns in humans. bioRxiv, 886879. bioRxiv
- Weghorn*, D., Balick*, D. J., Cassa, C., Kosmicki, J., Daly, M. J., Beier, D. R. & Sunyaev, S. R. (2019). Applicability of the mutation-selection balance model to population genetics of heterozygous protein-truncating variants in humans. Molecular Biology and Evolution, 433961. Journal
- Cassa*, C. A., Weghorn*, D., Balick*, D. J., Jordan*, D. M., Nusinow, D., Samocha, K. E., O'Donnell-Luria, A., MacArthur, D. G., Daly, M. J., Beier, D. R. & Sunyaev, S. R. (2017). Estimating the selective effects of heterozygous protein-truncating variants from human exome data. Nature Genetics, 49(5), 806. Journal PMC PubMed
Evolution of nucleosome positioning
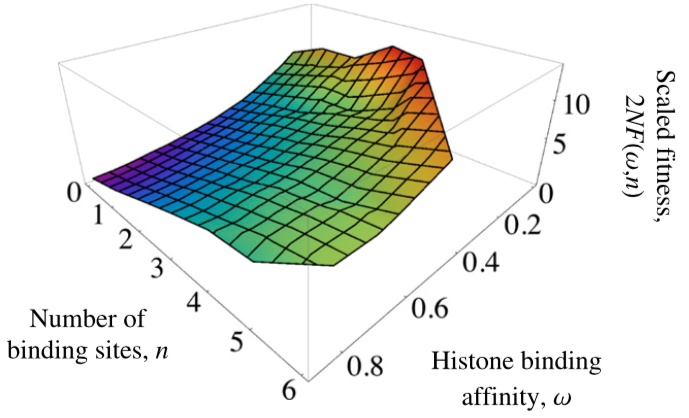
- Weghorn, D. & Lässig, M. (2013). Fitness landscape for nucleosome positioning. Proceedings of the National Academy of Sciences, 110(27), 10988-10993. Journal

Group Leader
Postdoctoral Researchers
Postdoctoral Researchers
Cancer Bayesian Selection Estimation (CBaSE)
We have developed a tool which derives gene-specific estimates of the strength of negative and positive selection in cancer. It accounts for the heterogeneity of mutation rate across the cancer genome, independent of external input information on mutation rate covariates. CBaSE estimates also take into account the context-dependent cancer type-specific mutational signature.
The CBaSE method and results were published in Weghorn & Sunyaev, Nature Genetics, 2017:
https://www.nature.com/articles/ng.3987.epdf
The CBaSE software can be downloaded as a standalone tool or used in a browser-based application here:
http://genetics.bwh.harvard.edu/cbase/index.html