 Novoa Lab
Novoa Lab
Genome Biology
- Group page
- Research lines
- Group members
- Publications


![]()
2024 - ICREA Research Professor and Group Leader, Centre for Genomic Regulation (CRG) - Spain
2018 - Group Leader, Centre for Genomic Regulation (CRG) - Spain
2016 - DECRA Senior Postdoctoral Fellow at Garvan Institute of Medical Research - Australia
2014 - HSFP Postdoctoral Fellow, Massachusetts Institute of Technology (MIT) and Broad Institute of MIT and Harvard - USA
2013 - EMBO Postdoctoral Fellow, Massachusetts Institute of Technology (MIT) and Broad Institute of MIT and Harvard, Boston - USA
2012 - Ph.D. in Biomedicine, Institute for Research in Biomedicine (IRB) - Spain
2009 - M.Sc. in Bioinformatics, University Pompeu Fabra (UPF) - Spain
2007 - B.Sc. in Biochemistry, University of Barcelona (UB) - Spain
*** Visit our lab website here: https://novoalab.com/ ***
Job Opportunities
Other positions
The Novoa Lab is always interested to hear from potential postdoctoral researchers, PhD students and technicians/bioinformaticians that are interested in joining our lab. Our laboratory is highly multidisciplinary, combining both wet and dry lab approaches, and therefore the recommended backgrounds range from biological sciences (e.g. Molecular Biology, Biochemistry, Biotechnology) to computational sciences (e.g. Computational Biology, Bioinformatics). Prior experience in RNA Biology, Nanopore sequencing or Machine Learning is a plus. If you are excited by our research and would like to join our team, please send an us an email (eva.novoa@crg.eu) with your CV and motivation letter.
News
Antibody design and cancer liquid biopsy research projects backed by ERC (11/07/2024)
Dr. Luis Serrano and Dr. Eva Novoa, researchers at the Centre for Genomic Regulation (CRG) in Barcelona, have each received a prestigious Proof of Concept (PoC) grant from the European Research Council in the first round of the 2024 competition.
EMBO selects Eva Novoa as new Young Investigator (17/01/2024)
The EMBO Young Investigator Programme is an initiative that supports excellent young group leaders in the early stages of their independent careers.
tRNA biomarkers for cancer diagnosis and prognosis enabled by new method (06/04/2023)
Researchers have developed a new method to measure tRNA abundances and their modifications in a simple, cost-effective manner, publishing their proof-of-concept findings in Nature Biotechnology.
Heart toggles between maintenance and energy-boost mode using ribosomes (07/03/2023)
Researchers at the CRG have discovered a mechanism involving ribosomes which helps the heart toggle between a ‘regular maintenance mode’ for day-to-day function and an ‘energy-boost mode’ which aids recovery for high-demand situations including heart attacks.
Researchers at the CRG receive €4.5m to study cancer, reproduction and blood formation (10/01/2022)
Eva Novoa, Group Leader of the Epitranscriptomics and RNA Dynamics lab at the CRG, will study the role of RNA in sperm in passing paternal hereditary information, for example through diet.
Two new grants awarded to the lab (October 2021)
The AECC (Spanish Association Against Cancer), under the LAB AECC 2021 call, has awarded the project “Native RNA nanopore sequencing as a novel technology for rapid cancer screening and monitoring” 300,000 € (role PI). The lab has also been awarded the Merck Innovation Grant H2020, amounting to 900,000 €, to the project “A drug discovery targeting cancer-specific RNA modifying enzimes” (role: co-PI). Both grants will run from 2021 to 2024.
Measuring RNA modifications opens new research avenues for cancer detection (14/05/2021)
Researchers at the Centre for Genomic Regulation (CRG) have developed a new method to measure the abundance of RNA modifications in much finer detail than previously possible.
Direct RNA sequencing made applicable to patient-derived samples (09/09/2020)
Researchers have developed a method to detect diverse RNA molecules, such as viral RNAs, in samples with minimal biological material.
New therapeutic targets for infertility and cancer revealed (08/05/2020)
CRG researchers share the result of the most comprehensive evolutionary analysis of RNA modification proteins to date.
Our Genome Biology paper highlighted in the media (El Diario, 07/05/20: https://www.eldiario.es/sociedad/Descubren-pueden-dianas-terapeuticas-infertilidad_0_1024698012.html)
CRG standardises COVID-19 data analysis to aid international research efforts (27/03/2020)
Researchers from the Centre for Genomic Regulation (CRG) have launched a new database to advance the international research efforts studying COVID-19.
Our efforts to create a SARS-CoV-2 direct RNA sequencing analysis repository have been highlighted in the press (Diari Ara, 27/03/20: https://www.ara.cat/societat/Barcelona-genetiques-coronavirus-covid-19_0_2424357698.html)
Further info related to these efforts can be found here.
Special feature of RNA modification detection methods in Science, including our work in bioRxiv (now published in Nature Comm) (Science, 17/05/2019: https://www.sciencemag.org/features/2019/05/epitranscriptomics-rna-revisited)
Summary
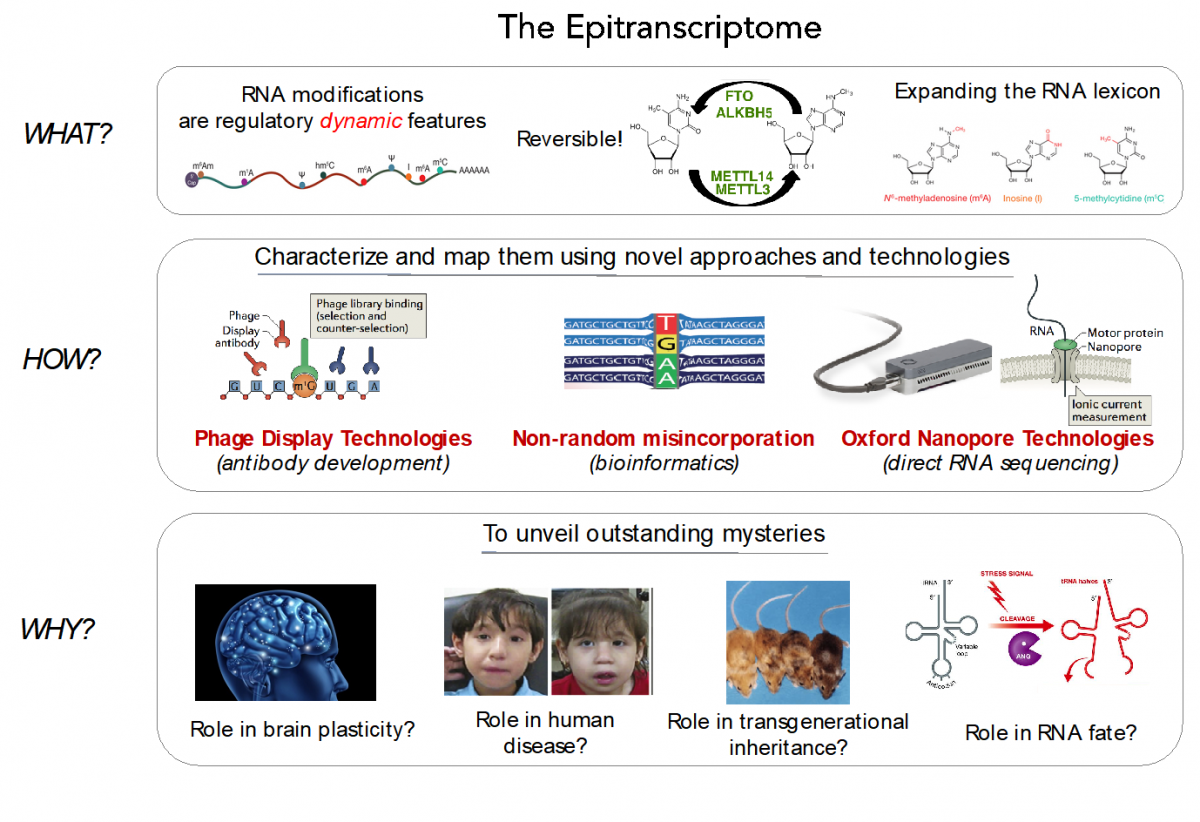
A current major challenge in biology is to understand how gene expression is regulated with surgical precision in a tissue-dependent, spatial and temporal dimension. Historically, genome-wide studies of gene expression have typically measured mRNA abundance rather than protein synthesis, in large part because such data are much easier to obtain. However, the correlation between mRNA levels and protein abundance is as low as r=0.35-0.40, suggesting that transcriptional regulation alone is not sufficient to unveil the complex orchestration of gene expression. In the last few decades, the scientific community has started to acknowledge the pivotal role that post-transcriptional regulatory mechanisms play in gene expression, however, we are still far from understanding how gene expression is finely tuned and regulated across tissues and conditions, suggesting that we are missing variables in the equation.
In our lab, we are employing a combination of experimental (RNASeq, polysome profiling, mouse/cell knockouts, Oxford Nanopore direct RNA sequencing) and computational techniques (NGS data analysis, algorithm development, machine learning), to unveil the secrets of three post-transcriptional regulatory layers: the epitranscriptome, RNA structure and ribosome specialization.
(Illustrations adapted from: Novoa et al., Nat Rev Mol Cell Biol 2017; Imanishi et al., Chem Communic 2017; Li et al., Nature Methods 2017; Hauenschild et al., Nucl Acids Res 2015; Stoecklin and Diederichs, EMBO J 2014)
Funding Acknowledgements
![]()
![]()
![]()




![]()

Proyecto PID2021-128193NB-I00 de investigación financiado por MICIU/AEI /10.13039/501100011033/ y por FEDER, UE
Our research lines include:
- Characterizing the role of RNA modifications (‘epitranscriptomics’) in human disease, synaptic plasticity and transgenerational inheritance
- Development of novel technologies and bioinformatics tools to map RNA modifications genome-wide, such as the use of direct RNA sequencing from Oxford Nanopore Technologies
- Single molecule RNA folding dynamics
- Characterization of the role, activity and regulators of specialized ribosomes in mammalian species
Research Lines
- Functional characterization of the unknown epitranscriptome
RNA modifications expand the RNA lexicon and are a fundamental component of RNA functionality. This code may not only enable or enhance specific chemistries in RNA-catalysed or RNA-dependent reactions, but also changes to RNA structure-function relationships that may alter signalling pathways, offering a layer of control to enable protein synthesis to be regulated in a space-, time- and signal-dependent manner.
Recently, advances in next-generation sequencing technologies have revealed the central role that these marks play in major cellular processes, such as splicing, cell fate transition or embryogenesis. Unfortunately, the limited availability of antibodies and chemicals selective to RNA modifications has so far limited our transcriptome-wide view to only a handful of RNA modifications. Consequently, the abundance, location, and function of the majority of RNA modifications remains unknown.
(Illustrations adapted from: Novoa et al., Nat Rev Mol Cell Biol 2017; Imanishi et al., Chem Communic 2017; Li et al., Nature Methods 2017; Hauenschild et al., Nucl Acids Res 2015; Stoecklin and Diederichs, EMBO J 2014)
In our lab, we are currently employing direct RNA sequencing from Oxford Nanopore Technologies (ONT), as a tool to produce high-resolution genome-wide maps of RNA modifications. In contrast to current methods used to detect RNA modifications genome-wide (RIP-Seq, Chem-Seq), ONT direct RNA sequencing is capable of detecting RNA modifications with single nucleotide resolution, in a quantitative manner, and in single molecules.
We aim to expand our understanding of the human epitranscriptome, and more specifically to decipher:
- their function and evolution
- the effects of their dysregulation in disease
- their roles brain plasticity
- their roles in transgenerational inheritance
Disentangling the role of RNA modifications in human disease
From the battery of over > 100 known RNA modifications, more than a dozen have already been linked to human diseases, including neurological disorders and cancer, highlighting their importance in proper cellular functioning. Several RNA modifications are highly enriched in brain, and mutations in several RNA methyltransferases have been associated with intellectual disability in humans and impaired cognitive ability. Unfortunately, the limited availability of antibodies and chemicals selective to RNA modifications has so far limited our transcriptome-wide view to only a handful of RNA modifications.
Using a combination of well-established cutting-edge experimental techniques, knockout mouse models and bioinformatics, we aim to analyse the distribution of certain modifications in human and mouse, to understand how dysregulation of RNA modifications lead to human diseases such as intellectual disability, cancer and infertility.

(Illustration from: Jonkhout N, Tran J, Schonrock N, Smith MA, Mattick JS and Novoa EM. The RNA modification landscape in human disease. RNA 2017)
- Development of novel technologies and bioinformatics tools to map RNA modifications genome-wide
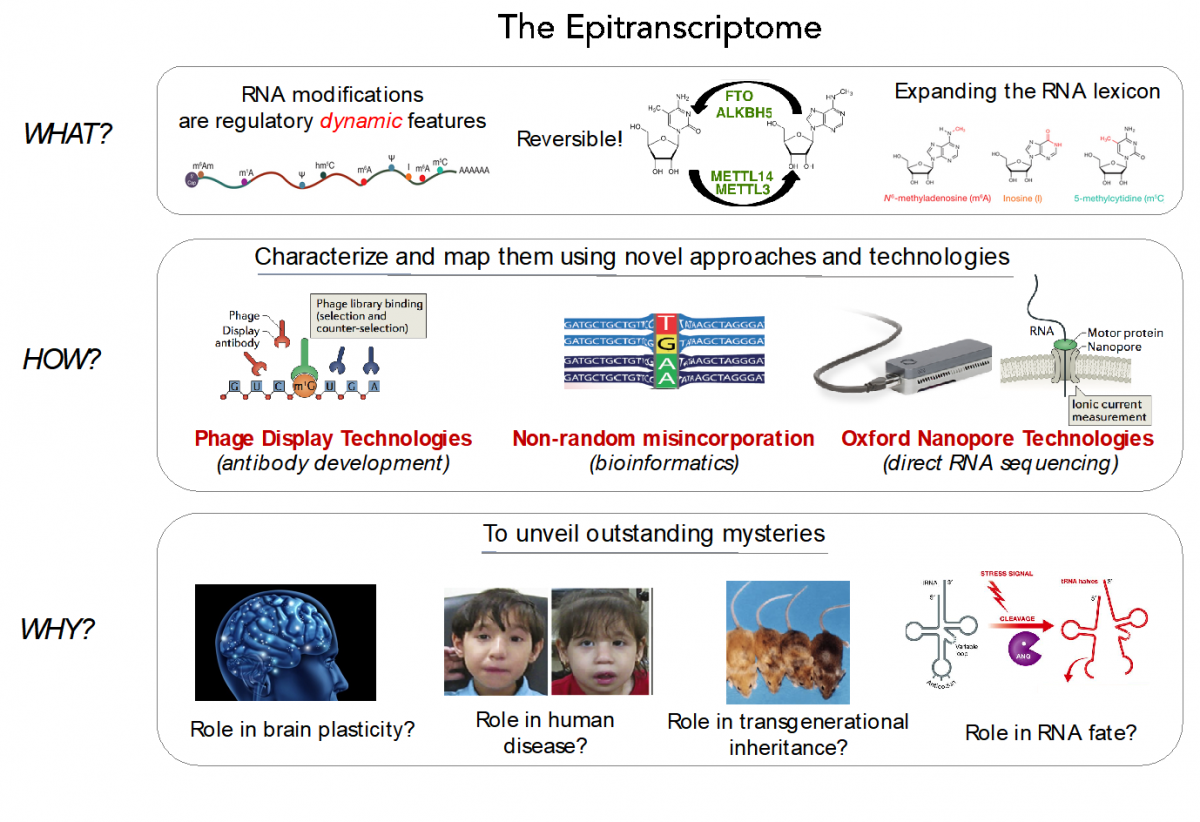
Currently, Sequencing-By-Synthesis (SBS) technologies, such as those developed by Illumina Corp., dominate genomic research. SBS platforms operate by DNA synthesis, copying a template strand, to cyclically incorporate individual fluorescently-labelled nucleotides that are captured with an optical device. Despite the major achievements of SBS technology, its major limitation is that it is typically blind to DNA and RNA modifications. Consequently, indirect methods (RIP-Seq, Chem-Seq) are required to identify RNA modifications genome-wide.
To overcome these limitations, we propose the application of two technologies to map RNA modifications:
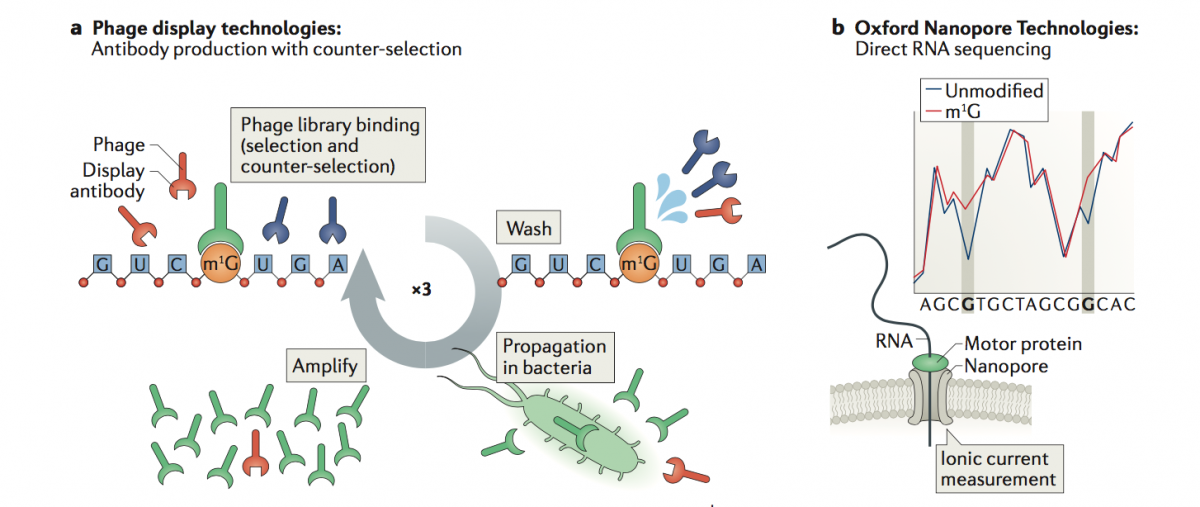
a) Phage Display Technologies (PDT) as a novel tool to produce antibodies against RNA modifications, where antibodies are generated by multiple rounds of antigen selection and competitor counter-selection.
b) Direct sequencing of RNA molecules using Oxford Nanopore Technologies (ONT), which employs thousands of membrane-embedded protein nanopores coupled to highly sensitive ammeters that measure ionic current passing through the pore. This technology avoids the reverse-transcription step during the library preparation, and thus can detect and measure RNA modifications from the RNA molecules that are being sequenced, in a quantitative manner, and with single nucleotide resolution.
To properly basecall RNA modifications from ONT output data, we are using a variety of tools, including machine learning, that can help us maximize the accuracy of detection of RNA modifications.
(Illustration from: Novoa EM, Mason CE, Mattick JS. Charting the unknown human epitranscriptome. Nature Rev Mol Cell Biol 2017)
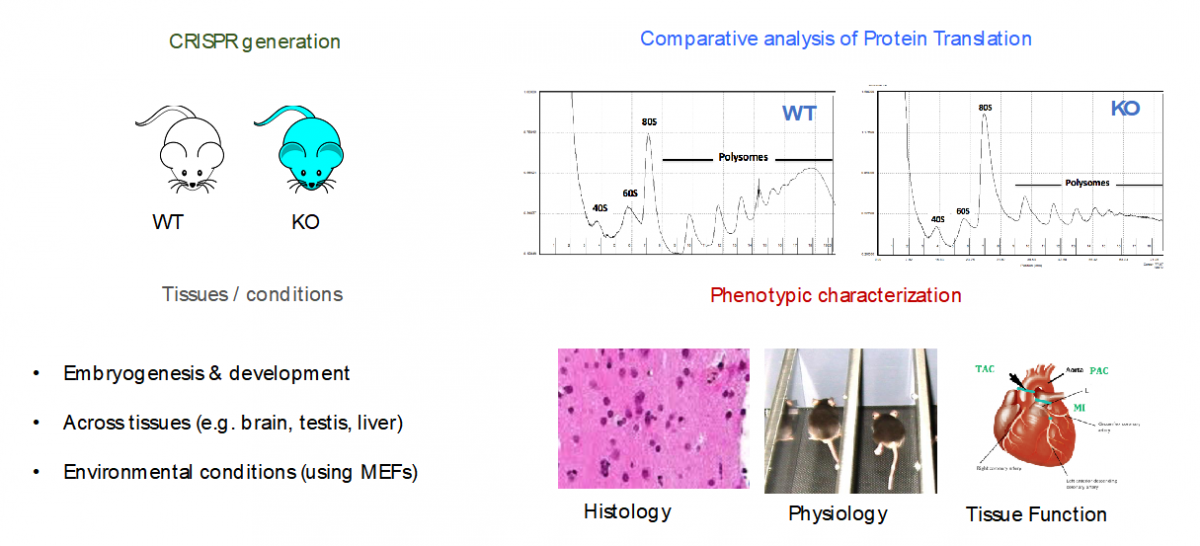
- Specialized ribosomes as a novel post-transcriptional regulatory layer to quickly tune the proteome
Ribosomes have been historically thought as uniform entities, however, recent evidence suggests that its composition might be dynamically regulated, and consequently, its activity (i.e., selective translation of subsets of transcripts).
In our lab, we are using a combination of molecular biology, mouse knockouts, genome-wide high-throughput techniques (i.e. RiboSeq, FracSeq, RNASeq) and bioinformatics, to decipher which are the forces that tune mammalian ribosome composition, and how its changes across tissues, developmental stages and environmental conditions, may affect cellular function.

Group Leader
Staff Scientist
Postdoctoral Researchers
PhD Students
Staff Scientist
Postdoctoral Researchers
PhD Students
![]()