 NEWS
NEWS
“Explainable” AI cracks secret language of sticky proteins
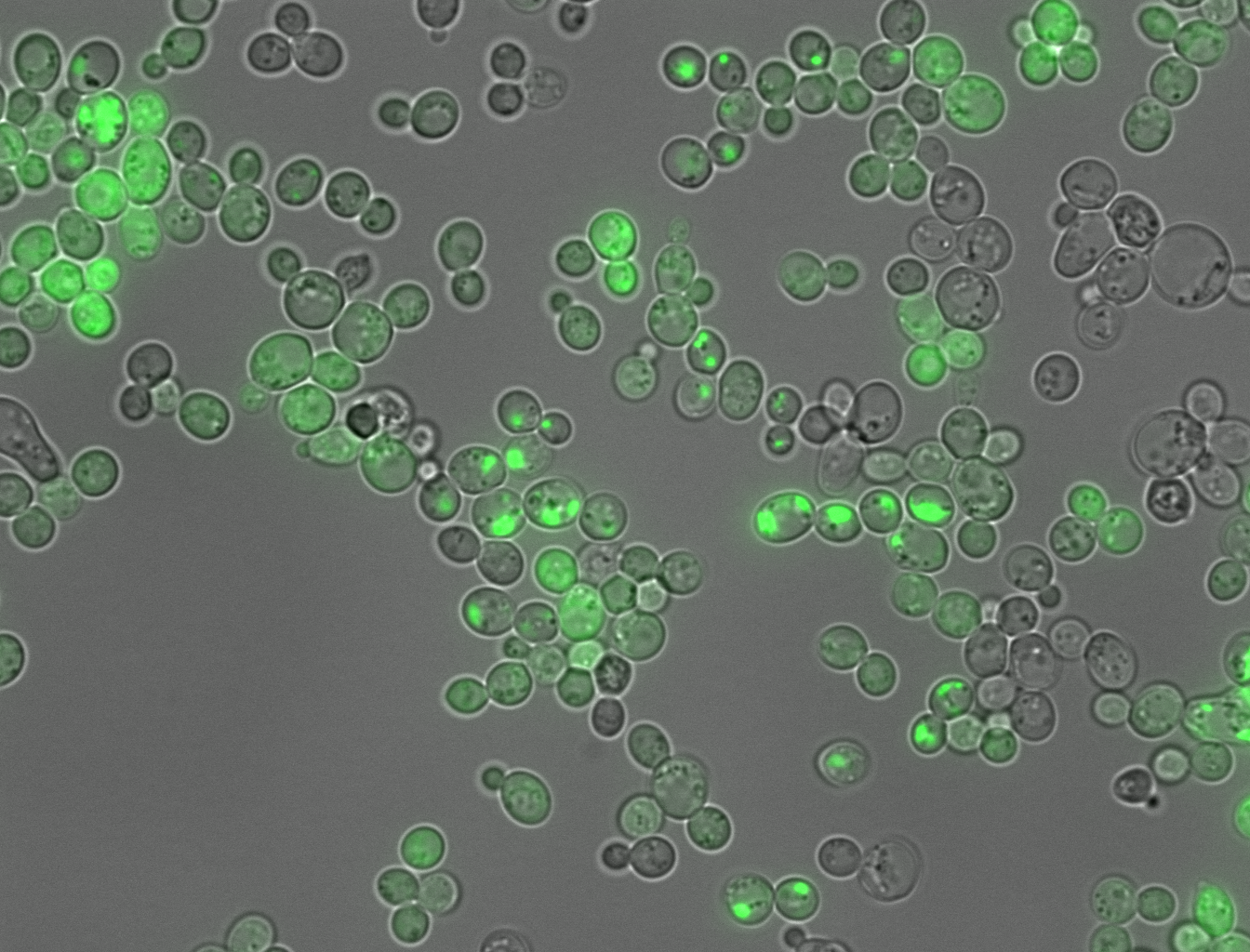
Amyloid aggregation inside cells. Credit: Benedetta Bolognesi/IBEC
An AI tool has made a step forward in translating the language proteins use to dictate whether they form sticky clumps similar to those linked to Alzheimer’s Disease and around fifty other types of human disease. In a departure from typical “black-box” AI models, the new tool, CANYA, was designed to be able to explain its decisions, revealing the specific chemical patterns that drive or prevent harmful protein folding.
The discovery, published today in the journal Science Advances, was possible thanks to the largest-ever dataset on protein aggregation created to date. The study gives new insights about the molecular mechanisms underpinning sticky proteins, which are linked to diseases affecting half a billion people worldwide.
Protein clumping, or amyloid aggregation, is a health hazard that disrupts normal cell function. When certain patches in proteins stick to each other, proteins grow into dense fibrous masses that have pathological consequences.
While the study has some implications for accelerating research efforts for neurodegenerative diseases, it’s more immediate impact will be in biotechnology. Many drugs are proteins, and they are often hampered by unwanted clumping.
“Protein aggregation is a major headache for pharmaceutical companies,” says Dr. Benedetta Bolognesi, co-corresponding author of the study and Group Leader at the Institute for Bioengineering of Catalonia (IBEC).
“If a therapeutic protein starts aggregating, manufacturing batches can fail, costing time and money. CANYA can help guide efforts to engineer antibodies and enzymes that are less likely to stick together and reduce expensive setbacks in the process,” she adds.
Protein clumps are formed using a poorly understood language. Proteins are made of twenty different types of amino acids. Instead of the usual A, C, G, T letters that make up the language of DNA, a protein’s language has twenty different letters, different combinations of which form “words” or “motifs”.
Researchers have long sought to decipher which combinations of motifs cause clumping and which others enable proteins to fold without error. Artificial intelligence tools that treat amino acids like the alphabet of a mysterious language could help identify the precise words or motifs responsible, but the quality and volume of data about protein aggregation needed to feed models have been historically scant or restricted to very small protein fragments.
The study addressed this challenge by carrying out large-scale experiments. The authors of the study created over 100,000 completely random protein fragments, each 20 amino acids long, from scratch. The ability for each synthetic fragment to clump was tested in living yeast cells. If a particular fragment triggered clump formation, the yeast cells would grow in a certain way that could be measured by the researchers to determine cause and effect.
Around one in every five protein fragments (21,936/100,000) caused clumping, while the rest did not. While previous studies might have tracked a handful sequences, the new dataset captures a much bigger catalogue of the different protein variants which can cause amyloid aggregation.
“We created truly random protein fragments including many versions not found in nature. Evolution has explored only a fraction of all possible protein sequences, while our approach helps us peer into a much bigger galaxy of possibilities, providing lots of data points to help understand more general laws of aggregation behaviour,” explains Dr. Mike Thompson, first author of the study and postdoctoral researcher at the Centre for Genomic Regulation (CRG).
The vast amount of data generated from the experiments was used to train CANYA. The researchers decided to create it using the principles of “explainable AI”, making its decision-making processes transparent and understandable to humans. This meant sacrificing a little bit of its predictive power, which is usually higher in “black-box” AIs. Despite this, CANYA proved to be around 15% more accurate than existing models.
Specifically, CANYA is a convolution-attention model, a hybrid tool borrowing from two distinct corners of AI. Convolution models, like those used in image recognition, scan photos for features like an ear or a nose to identify a face, except in this case CANYA skims through the protein chain to find meaningful features like motifs or “words”.
Attention AI models are used by language translation tools to identify key phrases in a sentence before deciding on the best translation. The researchers incorporated this technique to help CANYA figure out which motifs matter most in the grand scheme of the entire protein.
Together, these two approaches help CANYA see local motifs up close while also spotting their bigger-picture importance. The researchers could use this information to not just predict which motifs in the protein chain encourage clumping, block it, or something in between, but also understand why.
For example, CANYA showed that small pockets of water-repelling amino acids are more likely to spark clumping, while some motifs have a bigger impact on clumping if they’re near the start of a protein sequence rather than at the end. The observations align with previous findings researchers have seen under the microscope in known amyloid fibrils.
But CANYA also found new rules driving protein aggregation. For instance, certain building blocks of proteins, so-called charged amino acids, are normally thought to prevent clumping. But it turns out that in the context of other specific building blocks, they can actually promote clumping.
In its current form, CANYA primarily explains protein aggregation in yes or no terms, i.e. it works as a so-called “classifier”. The researchers next want to refine the system so it can predict and compare aggregation speeds rather than just aggregation likelihood. This could help predict which protein variants form clumps quickly and which do so more slowly, a vital factor in neurodegenerative diseases where the timing of amyloid formation matters just as much as the fact that it happens at all.
“There are 1024 quintillion ways of creating a protein fragment that is 20-amino acids long. So far, we’ve trained an AI with just 100,000 fragments. We want to improve it by making more and bigger fragments. This is just the first step but our work shows it is possible to decipher the language of protein aggregation. This is incredibly important for our understanding of human disease but also to guide synthetic biology efforts” concludes Dr. Bolognesi.
“This project is a great example of how combining large-scale data generation with AI can accelerate research. It’s also a very cost-effective method to generate data,” says ICREA Research Professor Ben Lehner, co-corresponding author and Group Leader at the Centre for Genomic Regulation (CRG) and the Wellcome Sanger Institute.
“Using DNA synthesis and sequencing we can perform hundreds of thousands of experiments in a single tube, generating the data we need to train AI models. This is an approach we are applying to many difficult problems in biology. The goal is to make biology predictable and programmable,” he adds.
The study is a joint collaborative effort by ICREA Research Professor Ben Lehner’s lab at the Centre for Genomic Regulation (CRG) and Benedetta Bolognesi’s lab at the Institute for Bioengineering of Catalonia (IBEC). Researchers from Cold Spring Harbor Laboratory (CSHL) and Wellcome Sanger Institute also collaborated in the study. It was funded by ”La Caixa” Research Foundation, the European Research Council and the Spanish Ministry of Science and Innovation.
EN CASTELLANO
CANYA, la IA española que descifra el lenguaje secreto de las proteínas “pegajosas”
Una herramienta de inteligencia artificial ha permitido dar un importante paso en la traducción del lenguaje que utilizan las proteínas para dictar si forman agregados pegajosos, cuya presencia se relaciona con el alzhéimer y otros cincuenta tipos de enfermedades humanas. A diferencia de los típicos modelos de IA de "caja negra", la nueva herramienta, CANYA, se diseñó para poder explicar sus decisiones, revelando los patrones químicos específicos que impulsan o previenen la agregación dañina de las proteínas.
El descubrimiento, publicado en la revista Science Advances, ha sido posible gracias al mayor conjunto de datos sobre agregación de proteínas creado hasta la fecha. El estudio ofrece nuevos conocimientos sobre los mecanismos moleculares que causan la agregación, que está relacionada con enfermedades que afectan a 500 millones de personas en todo el mundo.
La aglomeración de proteínas, o agregación amiloide, es un peligro para la salud que altera la función normal de las células. Cuando ciertas partes de las proteínas se adhieren entre sí, estas se convierten en masas densas y fibrosas que tienen consecuencias patológicas.
Si bien el estudio tiene algunas implicaciones para acelerar los esfuerzos en la investigación de enfermedades neurodegenerativas, su impacto más inmediato será en la biotecnología. Muchos fármacos son proteínas y, a menudo, su función se ve obstaculizada por agregaciones no deseadas.
"La agregación de proteínas es un gran dolor de cabeza para las compañías farmacéuticas", afirma la Dra. Benedetta Bolognesi, coautora principal del estudio y líder de grupo en el Instituto de Bioingeniería de Cataluña (IBEC).
"Si una proteína terapéutica comienza a agregarse, los lotes de fabricación pueden fallar, lo que cuesta tiempo y dinero. CANYA puede ayudar a guiar los esfuerzos para diseñar anticuerpos y enzimas que tengan menos probabilidades de adherirse y reducir los contratiempos en el proceso", añade.
Las agregaciones proteicas se forman utilizando un lenguaje todavía poco conocido. Las proteínas están formadas por veinte tipos diferentes de aminoácidos. En lugar de las habituales letras A, C, G, T que componen el lenguaje del ADN, el lenguaje de una proteína tiene veinte letras diferentes, cuyas combinaciones forman "palabras" o "motivos".
Se ha intentado durante mucho tiempo descifrar qué combinaciones de motivos causan la agregación amiloide y qué otras permiten que las proteínas se plieguen sin errores. Las herramientas de inteligencia artificial que tratan los aminoácidos como el alfabeto de un idioma misterioso podrían ayudar a identificar las palabras o motivos específicos responsables, pero la calidad y el volumen de los datos sobre la agregación de proteínas necesarios para alimentar los modelos han sido históricamente escasos o se han restringido a fragmentos de proteínas muy pequeños.
El estudio ha abordado este reto mediante la realización de experimentos a gran escala. Los autores del trabajo crearon más de 100.000 fragmentos de proteínas completamente aleatorios desde cero, cada uno de 20 aminoácidos de largo. La capacidad de cada fragmento sintético para agregarse se probó en células de levadura vivas. Así, si un fragmento en concreto desencadenara la formación de agregados, las células de levadura crecerían de una manera particular que puede ser medida para determinar la causa y el efecto.
Alrededor de uno de cada cinco fragmentos de proteína (21.936/100.000) causó aglomeración, mientras que el resto no lo hizo. Si bien estudios anteriores han podido rastrear un puñado de secuencias, el nuevo conjunto de datos ha registrado un catálogo mucho mayor de las diferentes variantes de proteínas que pueden causar la agregación amiloide.
"Hemos creado fragmentos de proteínas aleatorios, incluidas muchas versiones que no se encuentran en la naturaleza. La evolución ha explorado solo una fracción de todas las secuencias de proteína posibles, mientras que nuestro enfoque nos ayuda a asomarnos a una galaxia mucho mayor de posibilidades, proporcionando una gran cantidad de puntos de datos para ayudar a comprender las leyes más generales del comportamiento de agregación", explica el Dr. Mike Thompson, primer autor del estudio e investigador postdoctoral en el Centro de Regulación Genómica (CRG).
La gran cantidad de datos generados a partir de los experimentos se utilizó para entrenar a CANYA. El equipo decidió crearla utilizando los principios de la "IA explicable", haciendo que sus procesos de toma de decisiones fueran transparentes y comprensibles para los humanos. Esto significó sacrificar parte de su poder predictivo, que suele ser mayor en las IA de "caja negra". A pesar de ello, CANYA demostró ser alrededor de un 15% más precisa que los modelos existentes.
En concreto, CANYA es un modelo de convolución-atención, una herramienta híbrida que toma prestado de dos áreas distintas de la IA. Los modelos de convolución, como los que se utilizan en el reconocimiento de imágenes, escanean las fotos en busca de características como una oreja o una nariz para identificar una cara. De manera equivalente, CANYA ojea la cadena de proteínas para encontrar características significativas como motivos o "palabras".
Por otro lado, las herramientas de traducción de idiomas utilizan los modelos de IA para identificar frases clave en una oración antes de decidir cuál es la mejor traducción. El equipo incorporó esta técnica para ayudar a CANYA a descubrir qué motivos son los más importantes a escala general de toda la proteína.
Juntos, estos dos enfoques ayudan a CANYA a ver de cerca los motivos locales y, al mismo tiempo, a detectar su importancia a gran escala. Se puede usar esta información no solo para predecir qué motivos en la cadena de proteínas fomentan la aglomeración, la bloquean o provocan un estadio intermedio, sino también para comprender por qué.
Por ejemplo, CANYA demostró que las pequeñas regiones de aminoácidos repelentes al agua son más propensas a provocar aglomeración, mientras que algunos motivos tienen un mayor impacto en la aglomeración si se encuentran hacia el inicio de una secuencia de proteínas en lugar de hacia el final. Estas observaciones se alinean con hallazgos previos que se han visto bajo el microscopio en fibrillas amiloides conocidas.
Pero CANYA también encontró nuevas reglas que dirigen la agregación de proteínas. Por ejemplo, se pensaba que ciertos componentes básicos de las proteínas, los llamados aminoácidos cargados, evitan la aglomeración. Pero resulta que, en el contexto de otros bloques de construcción específicos, en realidad pueden promover la aglomeración.
En su forma actual, CANYA explica principalmente la agregación de proteínas en términos de sí o no, es decir, funciona como un llamado "clasificador". Cómo trabajo futuro, el equipo quiere refinar el sistema para que pueda predecir y comparar las velocidades de agregación en lugar de solo la probabilidad de agregación. Esto podría ayudar a predecir qué variantes de proteínas forman agregados rápidamente y cuáles lo hacen más lentamente, un factor vital en las enfermedades neurodegenerativas en las que el momento de la formación de amiloide es tan importante como el hecho de que ocurra.
"Hay 1.024 quintillones de formas de crear un fragmento de proteína de 20 aminoácidos de largo. Hasta ahora, hemos entrenado una IA con solo 100.000 fragmentos. Queremos mejorar el proceso creando más fragmentos y más grandes. Aunque este es solo el primer paso, nuestro trabajo muestra que es posible descifrar el lenguaje de la agregación de proteínas. Esto es increíblemente importante para nuestra comprensión de las enfermedades humanas, pero también para guiar los esfuerzos de la biología sintética", concluye la Dra. Bolognesi.
"Este proyecto es un gran ejemplo de cómo la combinación de la generación de datos a gran escala con la IA puede acelerar la investigación. También se trata de un método muy rentable para generar datos", dice el profesor de investigación ICREA Ben Lehner, coautor principal del estudio y jefe de grupo en el Centro de Regulación Genómica (CRG) y el Instituto Wellcome Sanger.
"Usando la síntesis y secuenciación de ADN, podemos realizar cientos de miles de experimentos en un solo tubo, generando los datos que necesitamos para entrenar modelos de IA. Este es un enfoque que estamos aplicando a muchos problemas difíciles de la biología, con el objetivode que esta sea predecible y programable", añade Dr. Lehner.
El estudio es fruto de la colaboración entre el laboratorio del profesor de investigación ICREA Ben Lehner en el Centro de Regulación Genómica (CRG) y el laboratorio de Benedetta Bolognesi en el Instituto de Bioingeniería de Cataluña (IBEC). Equipos del Laboratorio Cold Spring Harbor (CSHL) y el Instituto Wellcome Sanger también colaboraron en el estudio. El trabajo ha recibido financiación de la Fundación de Investigación "la Caixa", el Consejo Europeo de Investigación y el Ministerio de Ciencia e Innovación.
EN CATALÀ
CANYA, la IA catalana que desxifra el llenguatge secret de les proteïnes "enganxoses"
Una eina d'intel·ligència artificial ha permès fer un important pas en la traducció del llenguatge que utilitzen les proteïnes per dictar si formen agregats enganxosos, la presència dels quals es relaciona amb l'alzheimer i altres cinquanta tipus de malalties humanes. A diferència dels típics models d'IA de "caixa negra", la nova eina, CANYA, es va dissenyar per poder explicar les seves decisions, revelant els patrons químics específics que impulsen o prevenen l'agregació nociva de les proteïnes.
El descobriment, publicat a la revista Science Advances, ha estat possible gràcies al conjunt més gran de dades sobre agregació de proteïnes creat fins ara. L'estudi ofereix nous coneixements sobre els mecanismes moleculars que causen l'agregació, que està relacionada amb malalties que afecten 500 milions de persones a tot el món.
L'aglomeració de proteïnes, o agregació amiloide, és un perill per a la salut que altera la funció normal de les cèl·lules. Quan certes parts de les proteïnes s'adhereixen entre si, aquestes es converteixen en masses denses i fibroses que tenen conseqüències patològiques.
Si bé l'estudi té algunes implicacions per accelerar els esforços en la recerca de malalties neurodegeneratives, el seu impacte més immediat serà en la biotecnologia. Molts fàrmacs són proteïnes i, sovint, la seva funció es veu obstaculitzada per agregacions no desitjades.
"L'agregació de proteïnes és un gran mal de cap per a les companyies farmacèutiques", afirma la Dra. Benedetta Bolognesi, coautora principal de l'estudi i líder de grup a l'Institut de Bioenginyeria de Catalunya (IBEC).
"Si una proteïna terapèutica comença a agregar-se, els lots de fabricació poden fallar, cosa que costa temps i diners. CANYA pot ajudar a guiar els esforços per dissenyar anticossos i enzims que tinguin menys probabilitats d'adherir-se i reduir els contratemps en el procés", afegeix.
Les agregacions proteiques es formen utilitzant un llenguatge encara poc conegut. Les proteïnes estan formades per vint tipus diferents d'aminoàcids. En lloc de les habituals lletres A, C, G, T que componen el llenguatge de l'ADN, el llenguatge d'una proteïna té vint lletres diferents, les combinacions de les quals formen "paraules" o "motius".
S'ha intentat durant molt de temps desxifrar quines combinacions de motius causen l'agregació amiloide i quines altres permeten que les proteïnes es pleguin sense errors. Les eines d'intel·ligència artificial que tracten els aminoàcids com l'alfabet d'un idioma misteriós podrien ajudar a identificar les paraules o motius específics responsables, però la qualitat i el volum de les dades sobre l'agregació de proteïnes necessàries per alimentar els models han estat històricament escassos o s'han restringit a fragments de proteïnes molt petits.
L'estudi ha abordat aquest repte mitjançant la realització d'experiments a gran escala. Els autors del treball van crear més de 100.000 fragments de proteïnes completament aleatoris des de zero, cadascun de 20 aminoàcids de llarg. La capacitat de cada fragment sintètic per agregar-se es va provar en cèl·lules de llevat vives. Així, si un fragment en concret desencadenés la formació d'agregats, les cèl·lules de llevat creixerien d'una manera particular que pot ser mesurada per determinar la causa i l'efecte.
Al voltant d'un de cada cinc fragments de proteïna (21.936/100.000) va causar aglomeració, mentre que la resta no ho va fer. Si bé estudis anteriors han pogut rastrejar un grapat de seqüències, el nou conjunt de dades ha registrat un catàleg molt més gran de les diferents variants de proteïnes que poden causar l'agregació amiloide.
"Hem creat fragments de proteïnes aleatoris, incloses moltes versions que no es troben a la natura. L'evolució ha explorat només una fracció de totes les seqüències de proteïna possibles, mentre que el nostre enfocament ens ajuda a endinsar-nos a una galàxia molt més gran de possibilitats, proporcionant una gran quantitat de punts de dades per ajudar a comprendre les lleis més generals del comportament d'agregació", explica el Dr. Mike Thompson, primer autor de l'estudi i investigador postdoctoral al Centre de Regulació Genòmica (CRG).
La gran quantitat de dades generades a partir dels experiments es va utilitzar per entrenar CANYA. L'equip va decidir crear-la fent servir els principis de la "IA explicable", fent que els seus processos de presa de decisions fossin transparents i comprensibles per als humans. Això va significar sacrificar part del seu poder predictiu, que sol ser més gran a les IA de "caixa negra". Malgrat això, CANYA va demostrar ser al voltant d'un 15% més precisa que els models existents.
En concret, CANYA és un model de convolució-atenció, una eina híbrida que manlleva de dues àrees diferents de la IA. Els models de convolució, com els que s'utilitzen en el reconeixement d'imatges, escanegen les fotos tot cercant característiques com una orella o un nas per identificar una cara. De manera equivalent, CANYA escaneja la cadena de proteïnes per trobar característiques significatives com motius o "paraules".
D'altra banda, les eines de traducció d'idiomes utilitzen els models d'IA per identificar frases clau en una oració abans de decidir quina és la millor traducció. L'equip va incorporar aquesta tècnica per ajudar CANYA a descobrir quins motius són els més importants a escala general de tota la proteïna.
Junts, aquests dos enfocaments ajuden CANYA a veure de prop els motius locals i, alhora, a detectar la seva importància a gran escala. Es pot fer servir aquesta informació no només per predir quins motius a la cadena de proteïnes fomenten l'aglomeració, la bloquegen o provoquen un estadi intermedi, sinó també per comprendre per què.
Per exemple, CANYA va demostrar que les petites regions d'aminoàcids repel·lents a l'aigua són més propenses a provocar aglomeració, mentre que alguns motius tenen un major impacte en l'aglomeració si es troben cap a l'inici d'una seqüència de proteïnes en lloc de cap al final. Aquestes observacions s'alineen amb troballes prèvies que s'han vist sota el microscopi en fibril·les amiloides conegudes.
Però CANYA també va trobar noves regles que dirigeixen l'agregació de proteïnes. Per exemple, es pensava que certs components bàsics de les proteïnes, els anomenats aminoàcids carregats, eviten l'aglomeració. Però resulta que, en el context d'altres blocs de construcció específics, en realitat poden promoure l'aglomeració.
En la seva forma actual, CANYA explica principalment l'agregació de proteïnes en termes de sí o no, és a dir, funciona com un anomenat "classificador". Com a treball futur, l'equip vol refinar el sistema perquè pugui predir i comparar les velocitats d'agregació en lloc de només la probabilitat d'agregació. Això podria ajudar a predir quines variants de proteïnes formen agregats ràpidament i quines ho fan més lentament, un factor vital en les malalties neurodegeneratives en què el moment de la formació d'amiloide és tan important com el fet mateix que passi.
"Hi ha 1.024 quintilions de formes de crear un fragment de proteïna de 20 aminoàcids de llarg. Fins ara, hem entrenat una IA amb només 100.000 fragments. Volem millorar el procés creant més fragments i més grans. Tot i que aquest és només el primer pas, el nostre treball mostra que és possible desxifrar el llenguatge de l'agregació de proteïnes. Això és increïblement important per a la nostra comprensió de les malalties humanes, però també per guiar els esforços de la biologia sintètica", conclou la Dra. Bolognesi.
"Aquest projecte és un gran exemple de com la combinació de la generació de dades a gran escala amb la IA pot accelerar la recerca. També es tracta d'un mètode molt rendible per generar dades", diu el professor de recerca ICREA Ben Lehner, coautor principal de l'estudi i cap de grup al Centre de Regulació Genòmica (CRG) i l'Institut Wellcome Sanger.
"Usant la síntesi i seqüenciació d'ADN, podem realitzar centenars de milers d'experiments en un sol tub, generant les dades que necessitem per entrenar models d'IA. Aquest és un enfocament que estem aplicant a molts problemes difícils de la biologia, amb l'objectiu que aquesta sigui predictible i programable", afegeix Dr. Lehner.
L'estudi és fruit de la col·laboració entre el laboratori del professor de recerca ICREA Ben Lehner al Centre de Regulació Genòmica (CRG) i el laboratori de Benedetta Bolognesi a l'Institut de Bioenginyeria de Catalunya (IBEC). Equips del Laboratori Cold Spring Harbor (CSHL) i l'Institut Wellcome Sanger també van col·laborar en l'estudi. El treball ha rebut finançament de la Fundació de Recerca "la Caixa", el Consell Europeu de Recerca i el Ministeri de Ciència i Innovació.