 454 Sequencing
454 Sequencing
The 454 sequencing technology is a high throughput sequencing technology based on large-scale pyrosequencing. Post-sequencing analysis tools are included with the system. CRG purchased an FLX sequencing instrument in 2008 which can run both the standard FLX and the new Titanium chemistry. A comparison between the GS FLX standard platform and the Titanium platform is provided in Table 1.
- Sequencing workflow
- Sample requirements for the construction of 1 library / for 1 experiment
- Multiplexing samples
- Data processing
- Post sequencing software applications
- Useful SFF Tool commands
| FLX Titanium |
|---|
| 1 million reads per run |
| 350-500 Mb per run |
| >400 nt reads; Modal length= 500 nt, avg. length= 400 nt |
| Q20 read length of 400 bases |
| Runtime: 9 hours |
Table 1: Properties of the 454 Titanium platform. Run output refers to optimal conditions and will depend on sample properties. Source: http://www.454.com
Sequencing workflow
Sequencing workflow as explained by Roche 454.
Sample requirements for the construction of 1 library / for 1 experiment
| Genomic sequencing | |
|---|---|
| Single reads | 3 μg genomic DNA |
| Paired end libraries (3 kb span size) | 10 μg genomic DNA |
| Paired end libraries (8 kb span size) | 20 μg genomic DNA |
| Transcript sequencing | |
|---|---|
| Single reads | 2 μg cDNA |
| Fosmid, cosmid, BAC sequencing | |
|---|---|
| Single reads | 3 μg genomic DNA |
| Paired end libraries (3 kb span size) | 10 μg genomic DNA |
| Amplicon sequencing | |
|---|---|
| Single reads | 100 ng per amplicon |
Table 2: Sample requirements. DNA should be dissolved in 1 x TE buffer in a maximum volume of 70 μl (genomic DNA, cDNA), or 20 μl (amplicons). Genomic and cDNA samples should be quantified with the PicoGreen assay.
Please contact Heinz Himmelbauer for further technical inquiries.
Multiplexing samples
Bead deposition gaskets physically divide the PTP device into smaller regions (2, 4, 8, 16 regions). This enables simultaneous sequencing of multiple samples on a single PTP, while maintaining the unique identity. Table 4 gives the gasket format options and reads per region for FLX Titanium.
| Gasket format (regions) | Reads/region, Titanium |
|---|---|
| 2 | 450,000 |
| 4 | 160,000 |
| 8 | 80,000 |
| 16 | 15,000 |
Table 4: Reads per region for FLX Titanium. Run output refers to optimal conditions and will depend on sample properties. Source : http://www.454.com
MID identifiers
MID (Multiplex Identifier) are 10 nt sequence tags that can be added to the design of the adaptors used for library preparation, between the sequencing key and the template-specific primer, to help determine the source of the read. Roche presently supports 11 different Titanium MID identifiers. The MID-tagged samples can be pooled for simultaneous amplification and sequencing in a single region. Each MID is recognized by the analysis software, allowing for automated grouping and analysis of MID-containing reads (not yet available for Titanium).
Data processing
After completion of a sequencing run, data processing is done using the GS Run Processor application. This involves all the steps leading to the conversion of the raw image data to base-called results. There are two main steps in data processing - image processing and signal processing. Figures 1 and 2 list the input and output as well as the basic processing steps involved in each stage of data processing.
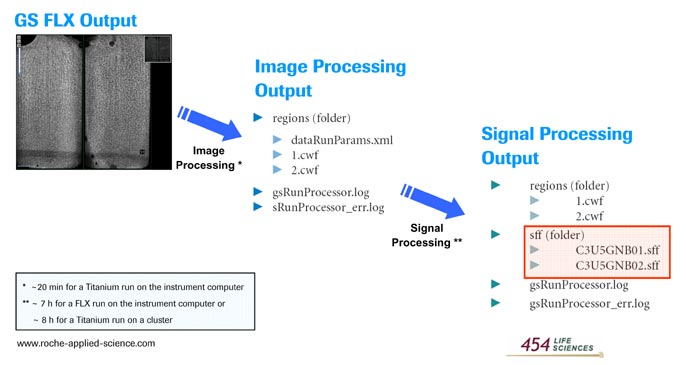
Figure 1: Summary of the image and signal processing pipeline
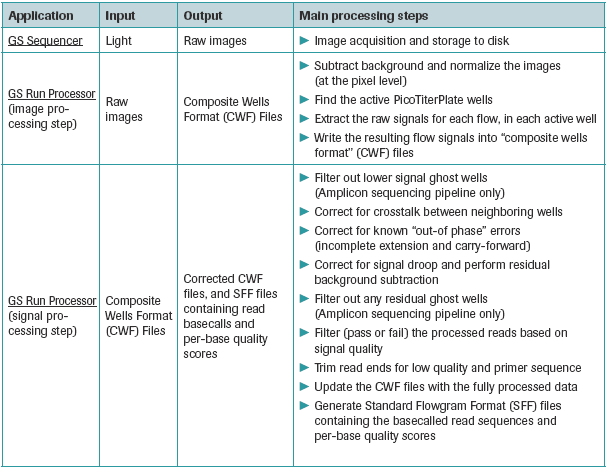
Figure 2: Roche-supplied post-sequencing applications. Source: 454 Genome Sequencer Data Analysis Software Manual
GS Sequencer: This application records a set of raw digital images representing the light detected over the PTP device, during each reagent flow of the sequencing run. This application is run in the sequencing instrument as it requires direct communication with the on-board camera and the microcontrollers. The output of this application includes the raw image files in .pif format.
Image processing: The image processing step extracts the raw signals for each nucleotide flow, in each active well in the PTP device from the raw images captured during the sequencing run. Image processing is performed real time, during the sequencing run as the images are being captured. The output flow signals for each region are written in a composite wells format (cwf) file. The cwf file is a "package" format which contains in addition to the uncorrected flowgrams metadata about the sequencing run and other intermediate metrics and data from the data processing.
Signal Processing: The signal processing step analyzes the signal data generated by the image processing step and stored in the .cwf files. Following a series of normalization, correction and quality filtering algorithms, it converts the remaining high quality signals into "flowgrams" for each read. The application generates basecalls and associated quality scores for the individual reads, and outputs this data as Standard Flowgram Format (SFF) files.
Information about reads which pass filters, as well metrics associated with each filter can be found in the two files "454BaseCallerMetrics" and "454QualityFilterMetrics". The base-called files for each region are outputted as FASTA files (.fna), while the associated quality files which contains the quality score for each base is outputted as .qual file.
Table 3 gives an approximate amount of disk space required to store the various types of files produced by the GS Run Processor application. The numbers assume that the bead loading gasket with the largest possible regions is used.
| Sequencing kit | Cycle number | Raw images | Raw images plus full processing |
|---|---|---|---|
| Titanium | 200 | 28 Gb | 37 Gb |
Table 3: Disk space required to store the various types of files produced by the GS Run Processor application. Source : 454 Software Manual
Post sequencing software applications
Figure 3 lists the three post sequencing software applications which are available to the user after the processed reads and their associated quality scores are obtained. It also lists the input and output of these applications, and gives an overview of the main processing steps involved in each application. Input for all these applications are SFF files
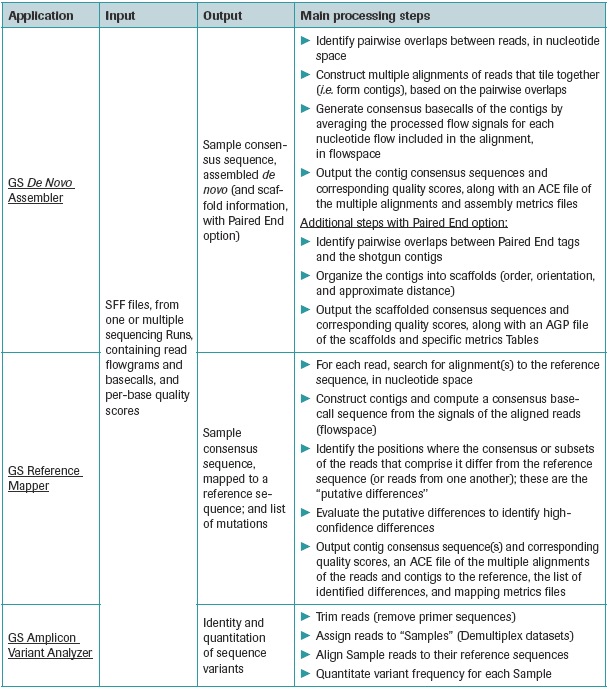
Figure 3: Brief outline of the post-sequencing software applications. Source : 454 Genome Sequencer Data Analysis Software Manual
GS De novo assembler (Newbler): This application assembles reads into contigs and generates a consensus sequence. The assembler also allows the inclusion of paired-end date into the analysis, enabling the ordering and orientation of the assembled contigs into scaffolds. The output of the assembler includes FASTA files of consensus basecalled contigs, corresponding quality files, metrics files providing various assembly metrics and ACE format files suitable for use in various sequence finishing programs.
GS Reference mapper: This application generates the consensus DNA sequence by mapping the reads to a reference sequence. It also generates a list of high confidence mutations. The read information in the SFF files serve as an input for this application. The output of the mapper includes FASTA files of consensus basecalled contigs, corresponding quality files, metrics files providing various mapping metrics, a text file listing the differences between the reference sequence(s) and the reads included in the mapping and ACE format files.
GS Amplicon Variant Analyzer: This application compares reads from an amplicon library to corresponding reference sequences, and allows the users to detect, identify and quantify the prevalence of sequence variants
Useful SFF Tool commands
We list below some useful SFF tool commands :
sfffile [options] [MIDlist@] (sfffile| datadir) ..
Modification(s) of sff file(s) (merging of two or more sff files, excluding certain reads from a sff file)
sffinfo [options] [- | sfffile] [accno ..]
Information extraction from a sff file (e.g. generating FASTA and quality scores files)
Output is in text file format
sff2scf locationstring [outputfile]
Conversion of SFF files to SCF files (for Consed)