 NOTICIAS
NOTICIAS
A new method accelerates the mapping of genes in the “Dark Matter” of our DNA
- Scientists at the Centre for Genomic Regulation (CRG) in Barcelona, have developed a new method, which improved the most important catalogue of genes -GENCODE-, including characterization of new genes in the DNA “Dark Matter”.
- Their new method, published today in Nature Genetics, offers a way of mapping genes in a more accurate, faster and cheaper way.
The information in the sequence of the human genome has a paramount importance in biomedical research. However, the value of this information is very limited in absence of a detailed map of the genes encoded in the genome. The genes are the basic biological units responsible for the biological traits of organism. Detailed information already exists, on the genomic regions that contain the genes that code for proteins, but the information about non-coding DNA regions – also known as DNA “dark matter” – lags behind. Here are found poorly-understood genes called “long non-coding RNAs” (lncRNAs), which are amongst the most numerous of all, and have been linked to a variety of diseases.
In a paper published today in Nature Genetics, an international team of scientists led by researchers at the Centre for Genomic Regulation (CRG) in Barcelona, in collaboration with researchers at Cold Spring Harbor in New York, the Wellcome Trust Sanger Institute in Hinxton, and qGenomics in Barcelona, sheds new light on this topic. In order to better identify, map, and characterize those “dark matter” genes, they have developed a new method that improves throughput and accuracy of current methods, and applied it in human and mouse.
“98% of our DNA does not encode for proteins. These DNA regions contain thousands of uncharacterised non-protein-coding genes, but there is still a long way to go until we fully understand their functions and their roles in disease. Reaching this goal will require complete gene maps. Our method represents an important step in this direction,” explained Rory Johnson, CRG alumnus currently Principal Investigator at the University of Bern, and co-leader of this paper.
The key feature of the new method, named RNA Capture Long Seq (CLS), is that it focuses specifically on the non-coding regions of the genome, that are amplified and analysed using the most advanced sequencing. “In this way we could produce a detailed map of 3,500 long non-coding RNAs in human and mice (about 20% of all those known so far) As a result, we characterized the genomic features of long-non-coding RNAs to better understand how these genes work,” stated Julien Lagarde and Barbara Uszczynska, co-first authors and CRG researchers.
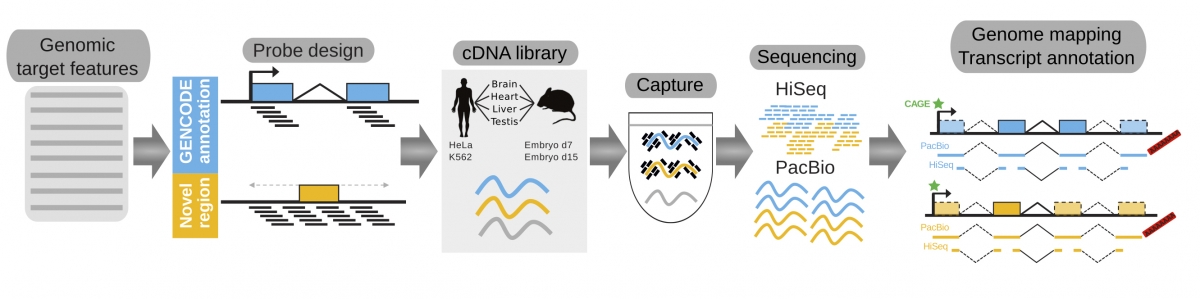
Researchers used this method to improve one of the most important genomic databases, GENCODE, which is the worldwide reference for the genes encoded in the human and mouse genomes. “Scientists around the globe are using GENCODE for their research projects as reference sets, so improving it means contributing to biomedical research worldwide”, said Roderic Guigó, coordinator of the Bioinformatics and Genomics Programme at the CRG and co-leader of this work. GENCODE was initiated in year 2003 by Roderic Guigó as part of the ENCODE ‘The Encyclopedia of the DNA elements’ project. Now, thanks to this new method, Guigó and collaborators have substantially improved the gene catalogues, in particular, for long non-coding RNA genes. “We have found a cheaper, faster and more accurate method that results in an improved catalogue and that will first benefit scientists worldwide and, ultimately, society,” concludes Guigó.
Almost 20 years after the Human Genome Project this work illustrates how our understanding on the information encoded in the genome is still evolving thanks to the development of increasingly powerful technologies. The better understanding of genomic function will lead, in turn, to a better understanding of health and disease.
Reference:
Julien Lagarde, Barbara Uszczynska-Ratajczak, Silvia Carbonell, Sílvia Pérez-Lluch, Amaya Abad, Carrie Davis, Thomas R Gingeras, Adam Frankish, Jennifer Harrow, Roderic Guigó and Rory Johnson. ‘High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing'. Nature Genetics. 2017. DOI: 10.1038/ng.3988
Funding information:
This work and its publication were supported by the National Human Genome Research Institute of the US National Institutes of Health (grants U41HG007234, U41HG007000 and U54HG007004) and the Wellcome Trust (grant WT098051). Rory Johnson was supported by the Ramón y Cajal Subprogram of the Spanish Ministry of Economy and Competitiveness (grant RYC- 2011-08851). Work in the laboratory of Roderic Guigó was supported by the National Human Genome Research Institute (awards U54HG0070, R01MH101814 and U41HG007234). This research was partly supported by NCCR RNA & Disease, funded by the Swiss National Science Foundation (to Rory Johnson). We acknowledge support from the Spanish Ministry of Economy and Competitiveness, Centro de Excelencia Severo Ochoa 2013–2017 (SEV-2012-0208), and from the CERCA Programme, Generalitat de Catalunya.
Media Contact:
Laia Cendrós, Media Relations, Centre for Genomic Regulation (CRG) - Tel.+34 93 316 02 37
| Adjunto | Tamaño |
|---|---|
| 107.23 KB | |
| 109.11 KB | |
| 106.35 KB |